

Migalastatest une thérapie orale innovante conçue pour le traitement de la maladie de Fabry, une maladie génétique rare du stockage lysosomal qui affecte plusieurs organes et réduit considérablement la qualité de vie. Contrairement aux thérapies enzymatiques de remplacement traditionnelles, le Migalastat agit en stabilisant certaines formes d’enzymes de l’organisme, lui permettant ainsi de fonctionner plus efficacement. Cet article fournit un aperçu complet et approfondi du Migalastat, y compris son mécanisme d'action, ses applications cliniques, ses avantages, ses limites, l'adéquation du patient, son profil de sécurité et son impact thérapeutique réel. Il est conçu pour aider les patients, les soignants et les acteurs de la santé à mieux comprendre comment cette médecine de précision remodèle les stratégies de gestion des maladies à long terme.
La maladie de Fabry est un trouble héréditaire rare du stockage lysosomal provoqué par des mutations du gène GLA, qui entraîne une activité déficiente ou absente de l'enzyme alpha-galactosidase A (α-Gal A). Ce déficit enzymatique entraîne l’accumulation de globotriaosylcéramide (Gb3) dans divers tissus, notamment les vaisseaux sanguins, les reins, le cœur et le système nerveux. Au fil du temps, cette accumulation entraîne des lésions organiques progressives, des douleurs chroniques, une insuffisance rénale, des maladies cardiaques et une réduction de l’espérance de vie.
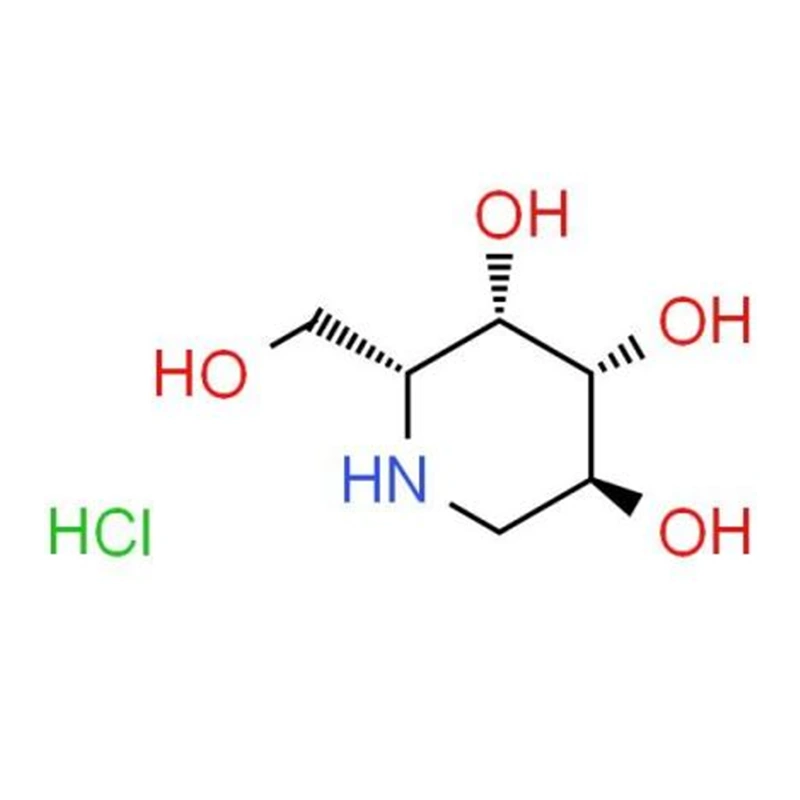
Le migalastat est un chaperon pharmacologique oral développé comme traitement ciblé pour les patients atteints de la maladie de Fabry présentant des mutations génétiques « sensibles » spécifiques. Au lieu de remplacer l’enzyme de manière externe, il stabilise l’enzyme du patient, l’aidant à se replier correctement et à atteindre les lysosomes où elle peut décomposer les substrats accumulés.
Ce mécanisme représente une avancée significative dans la médecine personnalisée, offrant une alternative à la thérapie enzymatique substitutive intraveineuse (ERT) à vie.
Le migalastat fonctionne comme un inhibiteur compétitif réversible qui se lie sélectivement à certaines formes mal repliées de l'alpha-galactosidase A. Ces mutations produisent souvent des enzymes structurellement instables mais néanmoins potentiellement fonctionnelles.
Son mécanisme peut être résumé en trois étapes clés :
Cette approche de « chaperon pharmacologique » garantit que les protéines enzymatiques endogènes sont sauvées au lieu d’être remplacées.
Résultat biologique clé :Réduction de l’accumulation de Gb3 et amélioration de la fonction cellulaire dans plusieurs organes.
Migalastatne convient pas à tous les patients atteints de la maladie de Fabry. Son efficacité dépend du fait que le patient présente ou non une mutation génétiquement « supportable ».
Les candidats idéaux incluent généralement :
Les tests génétiques sont essentiels avant de commencer le traitement, car seules des mutations spécifiques répondent efficacement au Migalastat.
Le migalastat est généralement administré sous forme de capsule orale prise tous les deux jours. Il doit être pris à jeun pour optimiser l'absorption.
| Paramètre | Détails |
|---|---|
| Formulaire | Gélule orale |
| Fréquence de dosage | Tous les deux jours |
| État d'administration | Estomac vide (2 heures avant ou après les repas) |
| Stockage | Température ambiante, endroit sec |
Les patients doivent respecter strictement les schémas posologiques pour maintenir des effets de stabilisation enzymatique constants.
Le Migalastat offre plusieurs avantages importants par rapport aux thérapies traditionnelles, notamment pour des profils génétiques adaptés.
Ces avantages améliorent considérablement l’observance et la qualité de vie des patients.
Malgré ses avantages, Migalastat présente plusieurs limites dont il faut tenir compte.
Ces contraintes soulignent l’importance du dépistage génétique personnalisé avant le début du traitement.
Le migalastat et la thérapie enzymatique substitutive (ERT) représentent deux approches distinctes de la prise en charge de la maladie de Fabry. Le tableau suivant présente leurs principales différences.
| Fonctionnalité | Migalastat | ERT (thérapie de remplacement enzymatique) |
|---|---|---|
| Voie d'administration | Gélule orale | Perfusion intraveineuse |
| Fréquence | Tous les deux jours | Toutes les 2 semaines |
| Mécanisme | Chaperon pharmacologique | Remplacement enzymatique |
| Éligibilité des patients | Mutations sensibles uniquement | Large applicabilité |
| Cadre clinique | À la maison | Hôpital/clinique |
Chaque thérapie a ses atouts et les décisions thérapeutiques dépendent du profil génétique, de la gravité de la maladie et des préférences du patient.
Le migalastat est généralement bien toléré, mais comme tous les médicaments, il peut provoquer des effets secondaires chez certaines personnes.
Les effets secondaires fréquemment rapportés comprennent :
Les effets indésirables graves sont rares mais nécessitent des soins médicaux s'ils surviennent. Une surveillance régulière de la fonction rénale et cardiaque est recommandée pendant le traitement.
Le parcours thérapeutique avec Migalastat commence par la confirmation génétique d’une mutation sensible. Une fois l’éligibilité confirmée, les patients passent à un plan de traitement structuré.
Une surveillance continue garantit que le traitement reste efficace et s'adapte à la progression de la maladie si nécessaire.
Des études cliniques ont démontré que le Migalastat peut améliorer ou stabiliser la fonction rénale, réduire la masse ventriculaire gauche dans le cœur et diminuer les biomarqueurs de la maladie chez les patients appropriés.
Des études observationnelles à long terme suggèrent une efficacité durable lorsque les patients respectent les protocoles de traitement. La recherche continue d'évaluer son rôle dans des stratégies plus larges de gestion de la maladie de Fabry.
Les essais cliniques en cours explorent également les thérapies combinées et les résultats à long terme en matière de protection des organes.
Q1 : Le Migalastat est-il un remède contre la maladie de Fabry ?
Non, le Migalastat n'est pas un remède. Il aide à gérer les symptômes et ralentit la progression de la maladie chez les patients éligibles.
Q2 : À quelle vitesse Migalastat agit-il ?
Des améliorations biochimiques peuvent être observées en quelques mois, mais les bénéfices cliniques peuvent prendre plus de temps en fonction de la gravité de la maladie.
Q3 : Les enfants peuvent-ils utiliser Migalastat ?
Les approbations actuelles se concentrent principalement sur les patients adultes, bien que des recherches soient en cours pour un usage pédiatrique.
Q4 : Que se passe-t-il si une dose est oubliée ?
Les patients doivent prendre la dose oubliée dès que possible, à moins qu'elle ne soit proche de la prochaine dose programmée.
Q5 : L’utilisation à long terme est-elle sûre ?
Les études à long terme indiquent une sécurité généralement favorable, mais une surveillance continue est nécessaire.
Le migalastat représente une avancée significative dans le traitement de la maladie de Fabry en offrant une option thérapeutique orale ciblée aux patients présentant des mutations génétiques spécifiques. Sa capacité à stabiliser la fonction enzymatique endogène offre une approche unique qui diffère fondamentalement des thérapies traditionnelles de remplacement enzymatique.
À mesure que la recherche progresse, le Migalastat pourrait jouer un rôle de plus en plus important dans les stratégies de médecine de précision pour les maladies génétiques rares. La surveillance clinique continue et l'innovation permettront d'affiner davantage ses applications et d'améliorer les résultats à long terme pour les patients.
Grâce à une sensibilisation croissante et à des capacités de dépistage génétique, davantage de patients pourraient avoir accès à cette thérapie, transformant ainsi la façon dont la maladie de Fabry est prise en charge à l'échelle mondiale.
Pour des solutions pharmaceutiques plus professionnelles, une collaboration en matière de recherche et une assistance en matière de formulation avancée liée aux thérapies contre les maladies rares, veuillez contacter :
Cosper Pharma Tech Co., Ltd.se spécialise dans le développement pharmaceutique de haute qualité et dans les technologies thérapeutiques innovantes conçues pour répondre aux besoins mondiaux en matière de soins de santé.
Si vous êtes intéressé par des opportunités de partenariat, des demandes de produits ou des conseils techniques, n'hésitez pas à nous contacter. Notre équipe est prête à accompagner votre projet du développement à la commercialisation.
Contactez-nousaujourd'hui pour explorer des solutions pharmaceutiques avancées avec Cosper Pharma Tech Co., Ltd.
![Ester 2-tétradécanoxyéthylique de l'acide 3-[3-imidazol-1-ylpropyl-[3-oxo-3-(2-tétradécanoxyéthoxy)propyl]amino]propanoïque](/upload/8820/3-3-imidazol-1-ylpropyl-3-oxo-3-2-tetradecanoxyethoxy-propyl-amino-propanoic-acid-2-tetradecanoxyethyl-ester-972329.webp "Ester 2-tétradécanoxyéthylique de l'acide 3-[3-imidazol-1-ylpropyl-[3-oxo-3-(2-tétradécanoxyéthoxy)propyl]amino]propanoïque")
-OH")
-cyclopropylglycine")


-OH")


 est-il suffisamment robuste pour une dégradation forcée et un dépôt ANDA ?")


